

The Burré lab is interested in understanding the molecular mechanisms underlying neurological disorders, with a focus on the neuronal synapse. Specifically, we are investigating the dysfunction of synucleins, a protein family implicated in Parkinson’s Disease and other synucleinopathies, and neurological diseases termed SNAREopathies, which are diseases related to dysfunction of SNARE and SNARE-related proteins such as VAMP2, SNAP-25, Syntaxin-1 and Munc18-1/STXBP1, with the aim to utilize the knowledge gained to develop rationale strategies to delay or prevent pathogenic changes. To address these aims, we employ a variety of technologies, including biophysics, biochemistry, cell biology, imaging, and electrophysiology on mouse models of neuropathology and human tissue.
Areas of Investigation
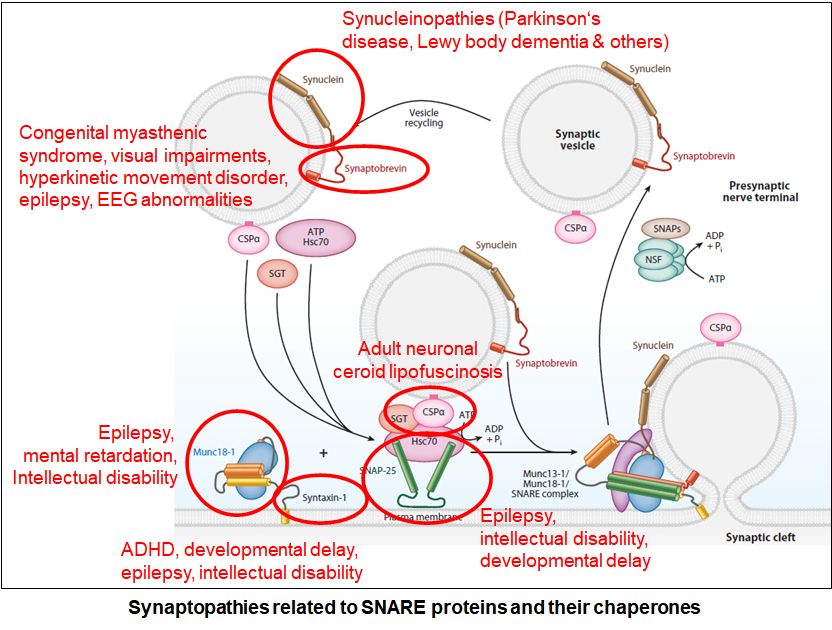
Function and Dysfunction of Synucleins at the Synapse
Aggregation of alpha-synuclein is the pathological hallmark of synucleinopathies, which include Parkinson’s disease, Lewy Body Dementia, and Multiple System Atrophy. In addition, alpha-synuclein aggregation is readily detectable in a variety of other degenerative diseases, including Alzheimer’s disease, Pick’s Disease, ALS, and Frontotemporal Dementia. Yet, despite 25 years of research, it remains enigmatic how alpha-synuclein triggers these devastating diseases. Our lab has several lines of investigations into synuclein function and dysfunction, including the identification of early disease biomarkers in mouse and human tissue and in the CNS and ENS, to better understand the physiological functions of α-, β- and γ-synuclein in in vitro models, culture models and mouse models, to determine the interplay and contribution of α-, β- and γ-synuclein to disease pathogenesis in in vitro models, culture models and mouse models, and to identify neuronal populations that are more vulnerable to disease spread across the brain.
STXBP1-linked diseases
Mutations in STXBP1 are associated with a growing list of syndromes, characterized by progressive cerebral dysfunction, leading to cognitive, sensory and/or motor function deterioration due to unremitting epileptic activity. STXBP1 is a neuronal protein which is essential for neurotransmitter release. Since 2008, more than 200 de novo mutations have been identified in the STXBP1 gene. We found mutations to trigger instability of STXBP1 and its interactors, resulting in synaptic dysfunction at multiple cellular levels. We also identified compounds to alleviate mutation-induced defects, one of which is in clinical trial with our clinical colleagues at Weill Cornell, the first trial for these devastating diseases. We are continuing to characterize the effects of mutations on neurons and expand our screen of disease-modifying therapies.
SNAP-25 encephalopathies
A growing list of de novo heterozygous mutations in the gene encoding the synaptic SNARE protein SNAP-25 lead to a disease that starts early in childhood and is characterized by unremitting epileptic activity combined with progressive cerebral dysfunction and cognitive, sensory and motor deterioration. Yet, the underlying disease mechanism remains unclear, and treatment strategies focus currently on symptom relief. We are studying 18 missense mutations in SNAP-25 regarding their effects on protein stability, subcellular localization, interactome, and neurotransmitter release. We are also currently testing novel compounds towards their effects on rescuing identified deficits.
VAMP2 encephalopathies
Similar to SNAP-25, heterozygous de novo mutations in the synaptic SNARE protein VAMP2/synaptobrevin-2 cause neurodevelopmental features including early onset axial hypotonia, intellectual disability, and features of autism spectrum disorder. We are taking the same approach as for SNAP-25, with the hope to identify therapeutic strategies to alleviate the identified pathogenic effects of the mutant protein.