
In a paper published Sept. 18 in Neuron, Dr. Li Gan, director of the Helen and Robert Appel Alzheimer’s Disease Research Institute in the Feil Family Brain and Mind Research Institute at Weill Cornell Medicine, and her colleagues describe how tau protein alters activity in a region of the neuron called the axon initial segment.
“Essentially what we discovered is that tau works as a thermostat to modulate neuronal activity,” said Dr. Gan, who is also the Burton P. and Judith B. Resnick Distinguished Professor in Neurodegenerative Diseases at Weill Cornell Medicine and the senior author of the study. “When tau is mutated, neuronal activity in the brain cannot be adjusted based on input and the system will become overactive and cause the hyperexcitability we see in frontotemporal dementia.”
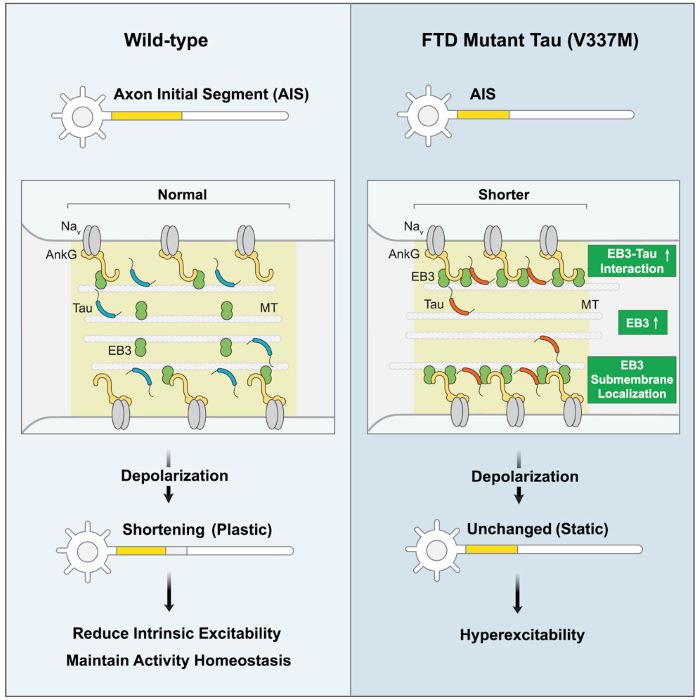
Neurons send electrical signals through axons to communicate with each other. In healthy neurons, the amount of electrical activity in the neuronal network are kept stable via a specialized structure on the axon called the axon initial segment. When neuronal activity becomes too high, the axon initial segment adjusts by shrinking in size in order to reduce the amount of electric activity in the brain. In dementia, there is evidence of elevated electrical signaling between neurons, even leading to seizures in some patients. Dr. Gan’s team discovered that axon initial segments in mutant neurons are the likely culprit due to their inability to dampen down activity when the inputs are high.
Using induced pluripotent stem cells (iPSCs)—which can differentiate into any cell type in the body—derived from patients carrying the tau mutation, the researchers created a human cell model of this process. They found that when tau was mutated, the axon initial segments of neurons did not shrink. This left the neurons unable to regulate activity, thereby becoming “overheated” or hyperexcited. The team verified these results by using CRISPR gene editing technology to introduce the tau mutation into normal human neurons grown in the lab.
“This shows us that the mutation is sufficient – not only necessary – to create this change,” said Dr. Gan, who is also the cofounder, stakeholder and consultant for Aeton Therapeutics. “Just this one change is enough to cause this altered neuronal activity.”
Dr. Gan and her collaborators at University California, Berkeley then used super-resolution imaging to look at what was happening at a molecular level in these cells. They saw that neurons with mutated tau had an abnormal distribution of a protein called EB3 in the axon initial segment. “We think this is why this structure becomes rigid and non-moveable,” said Dr. Peter Sohn, a research scientist in Dr. Gan’s team and the first author on the paper.
This understanding of the role of tau in neurons points toward the protein as a potential target for therapies that enhance cognitive function in people with FTD and Alzheimer’s disease, a more common dementia strongly linked with tau protein, Dr. Gan said. “One could potentially look for inhibitors of this abnormal interaction. If we can reduce the activity of pathogenic tau, we could improve the cognitive damages in patients suffering from these devastating conditions.”
The study is a collaboration between Weill Cornell Medicine investigators with researchers at Gladstone Institutes, UCSF, UC Berkeley and UC Santa Barbara.